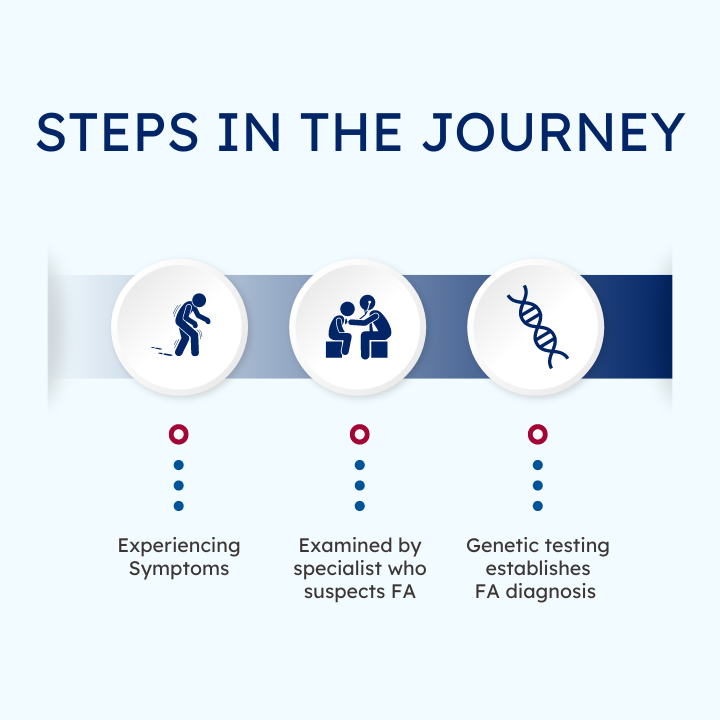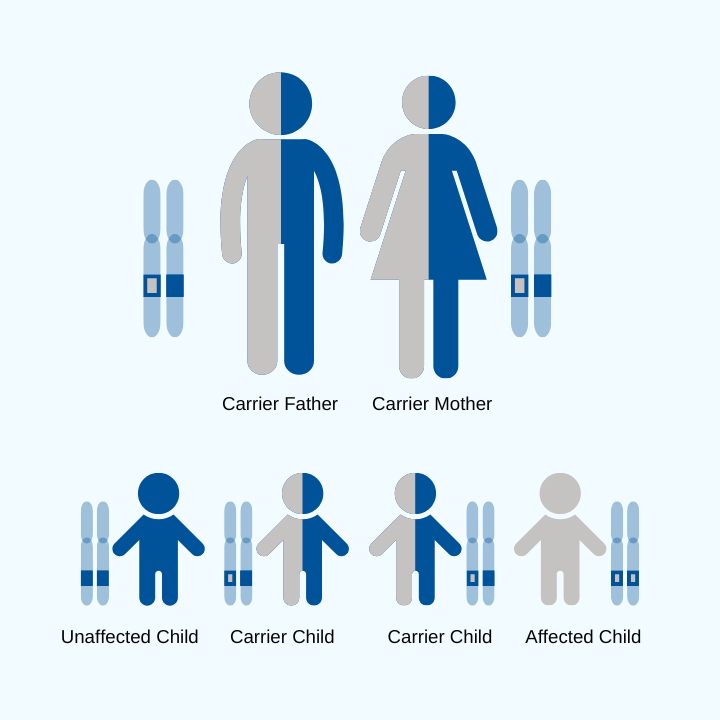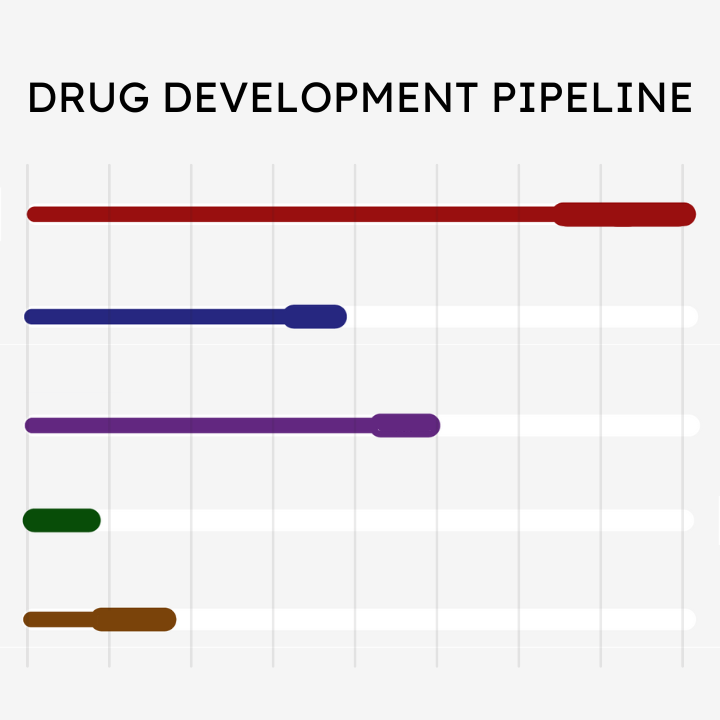







Friedreich’s ataxia can affect people in different ways. The age when symptoms first appear, often called age of onset, is associated with differences in how FA presents and progresses. Exact age of onset can be difficult to pinpoint, but it is generally defined as the age at which symptoms become noticeable enough to seek medical advice. Age of onset is related to the underlying genetics of FA. People with FA have expanded GAA repeats in both of their FXN genes. The smallest number of repeats between both genes is linked to age of onset, with more repeats on the smallest gene associated with earlier age of onset. In general, earlier onset may be associated with a more rapid progression, while later onset may be associated with slower progression of neurological symptoms. Our current understanding of how age of onset relates to symptoms and progression comes from natural history research.
Some individuals and families find it helpful to connect with others who have a similar age of onset, as it can provide shared understanding and support. If you’d like to be connected to other families living with FA, please reach out to us at: info@curefa.org or call the FARA office at (484) 879-6160.